Cystic Fibrosis (CF) is a multisystem genetic disorder that mainly affects the digestive and respiratory tract. The defective gene in CF causes the creation of thicker and stickier mucus that is difficult to remove/cough out of the lungs. This can lead to breathing difficulties and severe lung infections. The mucus can also interfere with pancreatic function by preventing the release of digestive enzymes from the pancreas that break down food. This can lead to malnutrition and poor growth in CF patients. Progressive lung disease is the major cause of death for most patients. Overtime, ranging from months to decades after birth, patients eventually develop a chronic infection in the respiratory tract with an array of bacterial flora, leading to progressive respiratory insufficiency and eventually respiratory failure. People with this life-threatening condition tend to have a shorter life span.
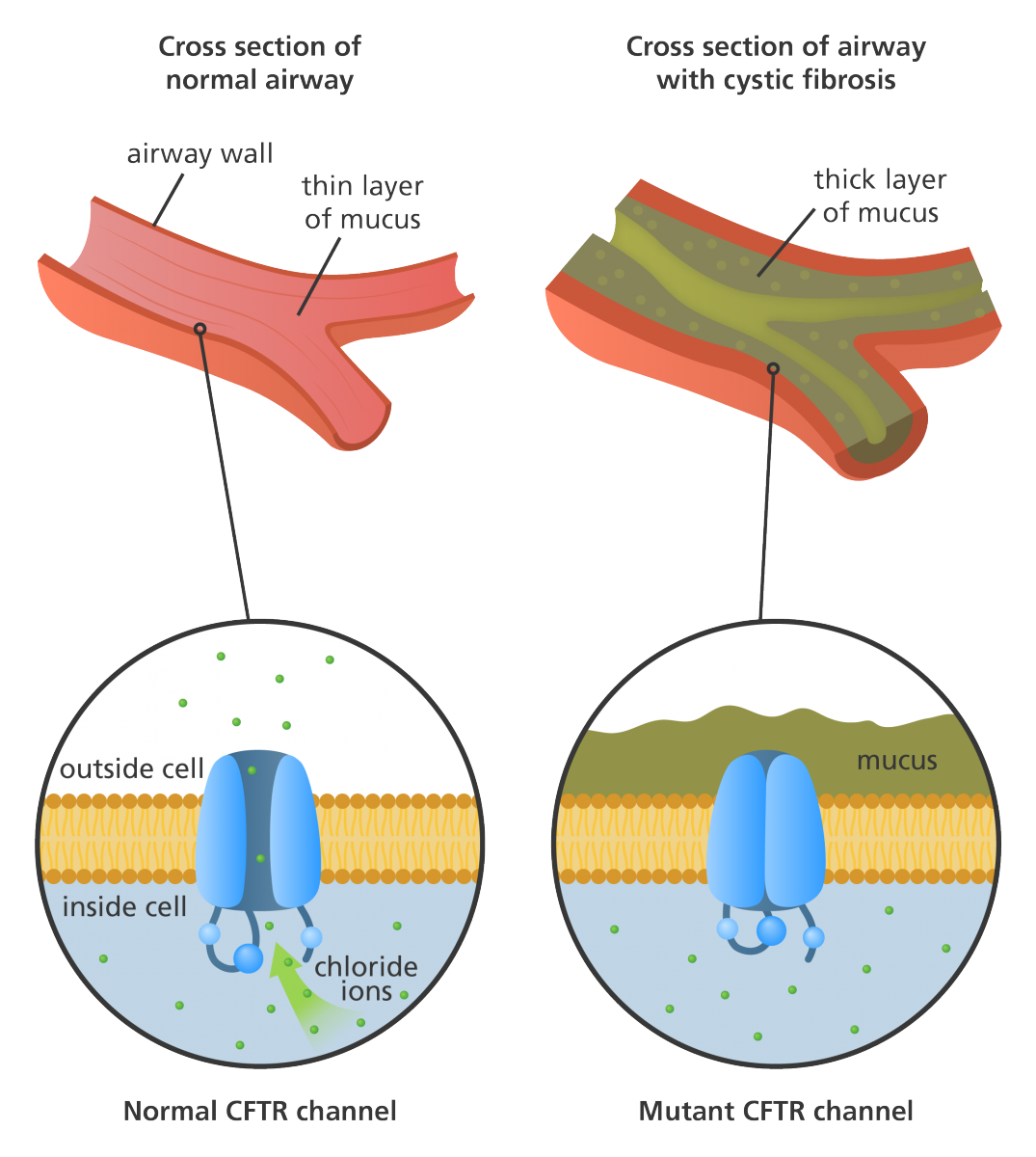
Patients with CF have abnormal transport of chloride and sodium across secretory epithelia which results in thickened secretions in the bronchi, biliary tract, pancreas, and intestines. This is due to the mutation in a single gene on chromosome 7 that encodes the cystic fibrosis transmembrane conductance regulator (CFTR) protein. Normally, CFTR regulates the transportation of chloride and sodium across the cell membrane. The transport of chloride ions from inside the cell to the cell surface helps attract water and thins the mucus. If the CFTR protein is dysfunctional the chloride ions are not allowed out of the mucus-producing cells to the surface and the mucus in various tubes, ducts and pathways will become thick and sticky which can obstruct those pathways. This obstructive process prevents bacteria from being cleared from the cells and can lead to infection. The types of complications in CF patients differ depending on the degree of mutation in the CFTR.
Many different types of bacterial pathogens colonize the lungs of patients with CF. Staphylococcus aureus (gram-positive) is usually the first pathogen to infect and colonize the airways leading to epithelial damage that then opens the way to other pathogens such as Pseudomonas aeruginosa (gram-negative).
1 P. aeruginosa has been shown to colonize in CF patients in more than 50% of cases and can survive for very long periods due to its ability to form biofilms.
1 Biofilm formation makes this bacterium difficult to treat with antibiotics. Burkholderia cepacia, a gram-negative bacterium found in the environment, can worsen lung disease in patients with CF.
2 It is not known how individuals with CF become infected with B. cepacia do not have a worsened lung condition while in other cases, B. cepacia can lead to a rapid decline in lung function and maybe death. B. cepacia can be passed from one individual with CF to another with CF, known as cross-infection. In the US, the ‘six feet apart’ rule exists stating that anyone with CF must stay at least six feet apart from another person with CF since more than just B. cepacia can be passed. Although in the UK, it is recommended that individuals with CF do not meet or spend time with each other. Nontuberculous mycobacteria (NTM) is another organism that can infect individuals with CF although the prevalence is much lower than those infected with S. aureus and P. aeruginosa. There is a stronger association between older individuals with CF and NTM.
3 NTM can cause progressive inflammatory lung disease however, NTM can also cause asymptomatic infections in CF patients.
The symptoms of CF are salty-tasting skin, persistent cough, shortness of breath, wheezing, hemoptysis, poor weight gain, bulky stools, and nasal polyps. Lung obstruction which creates optimal conditions for bacterial growth in CF increases the risk of lung infections such as pneumonia. Pancreas obstruction can lead to malnutrition and poor growth as well as osteoporosis.
References:
1. Coutinho, Henrique Douglas M et al. “Pulmonary bacterial pathogens in cystic fibrosis patients and antibiotic therapy: a tool for the health workers.” International archives of medicine vol. 1,1 24. 7 Nov. 2008, doi:10.1186/1755-7682-1-24
2. https://www.cff.org/Life-With-CF/Daily-Life/Germs-and-Staying-Healthy/What-Are-Germs/Burkholderia-Cepacia-Complex/
3. Floto RA, Olivier KN, Saiman L, et al. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis Thorax 2016;71:i1-i22.